
Tracheal Agenesis
2017
Tracheal agenesis is a rare, but fatal malformation with a prevalence of 1 in 50,00089 involving complete (8%) or partial absence of the trachea below the larynx. Approximately 53% of cases are associated with premature delivery. Often the proximal trachea ends in a blind sac just below the larynx with the lungs connected directly to the esophagus via the bronchi or a bronchoesophageal fistula.90 Classification of anatomic variations in this malformation is based on the length of the agenetic segment and the presence or absence of EA.77,80,90,91The most common variation is complete absence of the trachea below the larynx with the right and left main stem bronchi forming a common airway that is connected to the esophagus (56%). Other variations include (1) tracheal agenesis with fistulas connecting each main stem bronchi to the esophagus (10%); (2) agenesis of the proximal trachea with the main stem bronchi fused to form the distal trachea with (5%) or without (5%) a TEF; (3) a short segment of proximal trachea connected to the esophagus; (4) proximal and distal segments of the trachea, linked by a short atretic, or chord-like, fibrous band of tissue (5%); and (5) tracheal agenesis with an atretic strand joining the larynx to a distal tracheal segment with a fistula joining it to the esophagus (10%). As for EA/TEF, other congenital abnormalities are present in most cases, including additional lower respiratory tract malformations, such as abnormal lobe formation, and complex cardiac defects with a high incidence of abnormal vessels.
the website link is above on button.
Plus Type 3 by Floyd’s classification (Very Rare)
A rare but fatal congenital anomaly
In this report, we describe a newborn with a rare case of Type II tracheal agenesis and broncho-esophageal fistula. Polyhydramnios and suspected esophageal atresia were identified during routine prenatal ultrasound screening. Upon delivery, rigid bronchoscopy, esophagoscopy, and intraoperative fluoroscopy was performed, where both bronchi and the carina showed unusual horizontal orientation making it difficult to identify the fistula. However, a post-mortem CT confirmed the diagnosis of an isolated Type II tracheal agenesis with broncho-esophageal fistula. Tracheal agenesis is a rare and lethal congenital anomaly, where a complete interruption or absence of the trachea is present. Since it was initially described in 1900, few cases have been published worldwide. The prevalence of tracheal agenesis is less than 1:50,000 with a male to female ratio of 2:1. In general, 52% of cases are associated with premature delivery and approximately half of the cases are associated with polyhydramnios.
With Photos
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3277332/
Tracheal agenesis is a catastrophic congenital anomaly that invariably results in death. Forty-seven cases have been previously reported in the literature. We add five additional cases, including two type 1 cases, two type 2 cases, and one type 3 case, based on Floyd’s classification scheme. We describe the features of this unusual anomaly at the time of diagnosis. We discuss a rational approach to the management of this difficult problem on an emergent basis that allows for the maintenance of the infant’s life until all of the implications of this fatal condition can be assessed. While we do not advocate reconstructive surgery for this anomaly, which has been universally fatal, we discuss the potential rearrangement of the anatomy, which may offer some hope in future cases. The concomitant congenital anomalies associated with these cases are reviewed, and autopsy specimens are presented for their anatomic interest.
http://journals.sagepub.com/doi/pdf/10.1177/000348949210100704
Click here to read more
http://www.annalsthoracicsurgery.org/article/S0003-4975(10)63123-2/pdf
Tracheal agenesis is an extremely rare congenital anomaly involving the respiratory system. It is generally associated with anomalies of other systems. Antenatal diagnosis of this condition is difficult; therefore, it presents as a medical emergency in the labor room. Intubation in these babies is difficult. As many of these babies are born prematurely, respiratory distress syndrome (RDS) adds to the management difficulties. Here, we describe two babies with this lethal anomaly and RDS where esophageal intubation and surfactant therapy proved beneficial. Furthermore, described are other associated anomalies. A male baby was born to a 25-year-old mother at 30 weeks of gestation. The mother had presented in emergency with preterm labor and her antenatal ultrasonography (US) done at 26 weeks was suggestive of polyhydramnios. The baby was delivered vaginally and weighed 1.2 kg. As the baby did not cry immediately, endotracheal intubation was attempted to secure the airway but the vocal cords could not be visualized and there was a single opening corresponding to the esophagus.
The photo will enlarge if clicked on, (Evidence of respiratory distress syndrome)
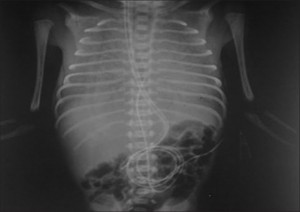
Resuscitation was hence continued with bag and mask ventilation and baby was shifted to the NICU where a second attempt was made to intubate which failed again. As the baby’s condition worsened, the endotracheal tube was passed in the visualized opening, after which the saturations improved. In view of X-ray chest, suggestive of severe respiratory distress syndrome (RDS), natural bovine surfactant with 100 mg/kg of phospholipids was given through the “endotracheal tube,” which showed improvement suggesting that a connection existed between the esophagus and trachea. On invasive ventilation with high-pressure support and 100% FiO2, the saturation was maintained at 85%. The baby also had an imperforate anus, a structurally normal heart on echocardiography, and normal abdominal US. Because of the critical condition of the baby, computed tomography (CT) was not attempted but a water-soluble dye was instilled through the endotracheal tube which showed the esophageal tracheal connection. The baby further got complicated with a developing pneumoperitoneum, which was drained, but the baby died at 48 h of life.
Much more on this with many x-rays
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4970349/
Ea-tef & Tof-oa related conditions
Thomas Richards survive’s birth without a trachea
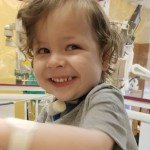 Our little man is pretty famous in our area of the globe. More or less area of our home state of Wisconsin. He’s had a few stories written about him. He is as far as we have been told and know the oldest survivor in the United States. The link provided is to his debut story written and explains in great detail about the beginning of our journey with TA. And this Saturday he will be 2 years old!! (2018)
Our little man is pretty famous in our area of the globe. More or less area of our home state of Wisconsin. He’s had a few stories written about him. He is as far as we have been told and know the oldest survivor in the United States. The link provided is to his debut story written and explains in great detail about the beginning of our journey with TA. And this Saturday he will be 2 years old!! (2018)
Deep in the womb, about the time that the human embryo is, in its entirety, about the size of a peppercorn, a tube sprout’s in the rapidly evolving realm of its primitive lungs. As the tube grows, two ridges form along its length inside. These ridges eventually fuse, creating two tubes. The one in front becomes the trachea — the windpipe — through which we breathe. The one behind becomes the esophagus – the gullet — through which we swallow. Mature, the trachea begins below the voice box, descends three or four inches, and then branches to the lungs. Poke the base of your neck, just above your chest, and you can feel the rings of its stiff, cartilaginous shell.
 The esophagus is a stretchy hose of muscle, twice the length of the trachea, that leads from the throat to the stomach. In more than a century, medical literature has recorded fewer than 200 cases in which the fetal trachea fails to form. Babies born with this anomaly, called tracheal agenesis, die silently, having never drawn a breath. Only five, and only due to extraordinary surgical intervention, have survived.
The esophagus is a stretchy hose of muscle, twice the length of the trachea, that leads from the throat to the stomach. In more than a century, medical literature has recorded fewer than 200 cases in which the fetal trachea fails to form. Babies born with this anomaly, called tracheal agenesis, die silently, having never drawn a breath. Only five, and only due to extraordinary surgical intervention, have survived.
None in the United States. None, until Thomas David Richards, born this spring at Ministry St. Joseph’s Children’s Hospital in Marshfield.
The team of doctors and nurses who treated Thomas Richards pose with him and his family shortly before his discharge from Children’s Hospital of Wisconsin on Sept. 1. They are (from left) Michael McCormick, Cecilia Lange, Mike Mitchell, Keith Oldham, John Densmore (holding Thomas), father Corey Richards, mother Jessica Richards, nurse Heather Sepp and child life specialist Molly McCormick.
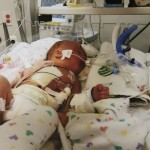 The medical team moved Thomas to an operating room, where a surgeon made an unsuccessful attempt to find Thomas’ trachea.
The medical team moved Thomas to an operating room, where a surgeon made an unsuccessful attempt to find Thomas’ trachea.
Thomas did not appear to have a trachea, she explained. What air was reaching him was flowing down his esophagus and through a tiny fistula, a passageway, that opened to his lungs. None of the three had ever seen such a thing. But, they decided, if they could get air to Thomas’ lungs by putting a breathing tube down his esophagus, then that’s what they would do. Thomas was placed on a ventilator. Almost immediately, Dominguez was presented with a new problem.
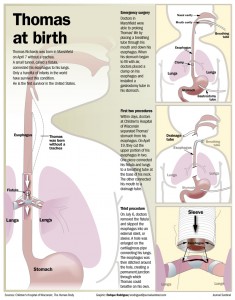 Most of the air ventilating through Thomas’ esophagus was ending up in his stomach. Some of it reached his lungs, but every breath the ventilator delivered expanded his stomach more, compressing the lungs the team was so furiously trying to inflate. Woods suggested that Dominguez clamp Thomas’ esophagus just above his stomach. That would keep air out and force it through the fistula. It was pure improvisation, and though Dominguez had never attempted such a procedure, Woods’ suggestion made sense.
Most of the air ventilating through Thomas’ esophagus was ending up in his stomach. Some of it reached his lungs, but every breath the ventilator delivered expanded his stomach more, compressing the lungs the team was so furiously trying to inflate. Woods suggested that Dominguez clamp Thomas’ esophagus just above his stomach. That would keep air out and force it through the fistula. It was pure improvisation, and though Dominguez had never attempted such a procedure, Woods’ suggestion made sense.
Dominguez reached Thomas’ esophagus through an incision in his abdomen. She had to simply guess at how firmly she should close the clamp. Too tight, and the tissue would fall apart. Too loose, and the clamp would let air leak through or fall off. She gave it her best shot, then installed a gastrostomy tube — a feeding tube — through the same incision to deflate his stomach.
She then affixed a patch over the incision and, about 9 p.m., sent Thomas by ambulance to Children’s Hospital. She went home, exhausted but unable to sleep, wondering if Thomas would survive the three-hour journey.
The Full Story on this brave Child can be found here look below.
https://www.jsonline.com/story/news/health/2016/11/12/days-life-thomas-richards-story/93423880/
Children’s Hospital of Wisconsin
https://www.childrenshospitals.org/About-Us/About-Childrens-Hospitals
Twin pregnancy complicated by esophageal atresia, duodenal atresia
Twin pregnancy complicated by esophageal atresia, duodenal atresia, gastric perforation, and hypoplastic left heart structures in one twin: a case report and review of the literature.
Published: 18 March 2017
This is very long with x-rays, Graphs, etc well worth reading so click on the link below
Background
The antenatal diagnosis of a combined esophageal atresia without tracheoesophageal fistula and duodenal atresia with or without gastric perforation is a rare occurrence. These diagnoses are difficult and can be suspected on ultrasound by nonspecific findings including a small stomach and polyhydramnios. Fetal magnetic resonance imaging adds significant anatomical detail and can aid in the diagnosis of these complicated cases. Upon an extensive literature review, there are no reports documenting these combined findings in a twin pregnancy. Therefore we believe this is the first case report of an antenatal diagnosis of combined pure esophageal and duodenal atresia in a twin gestation.
Case presentation
We present a case of a 30-year-old G1P0 white woman at 22-weeks gestation with a monochorionic-diamniotic twin pregnancy discordant for esophageal atresia, duodenal atresia with gastric perforation, hypoplastic left heart structures, and significant early gestation maternal polyhydramnios. In this case, fetal magnetic resonance imaging was able to depict additional findings including the area of gastric wall rupture, hiatal hernia, dilation of the distal esophagus, and area of duodenal obstruction and thus facilitated the proper diagnosis. After extensive counseling at our multidisciplinary team meeting, the parents elected to proceed with radiofrequency ablation of the anomalous twin to maximize the survival of the normal co-twin. The procedure was performed successfully with complete cessation of flow in the umbilical artery and complete cardiac standstill in the anomalous twin with no detrimental effects on the healthy co-twin.
Conclusions
Prenatal diagnosis of complex anomalies in twin pregnancies constitutes a multitude of ethical, religious, and cultural factors that come into play in the management of these cases. Fetal magnetic resonance imaging provides detailed valuable information that can assist in management options including possible prenatal intervention. The combination of a cystic structure with peristalsis-like movement above the diaphragm (for example, “the upper thoracic pouch sign”), polyhydramnios, and progressive distention of the stomach and duodenum should increase suspicion for a combined pure esophageal and duodenal atresia.
This is much longer than I have put here.
https://jmedicalcasereports.biomedcentral.com/articles/10.1186/s13256-016-1195-x
.
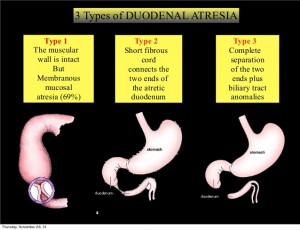
Duodenal Atresia
What is duodenal atresia?
The duodenum is the first portion of small intestine after the stomach that has many connections to and shares blood vessels with other organs such as the liver, gallbladder, and pancreas. When part of the bowel fails to develop normally in the fetus, a blockage of the duodenum can occur, otherwise known as an atresia or bowel obstruction. Duodenal atresias can occur as a complete or partial blockage of any portion of the duodenum. Newborns diagnosed with duodenal atresia often present with vomiting.
Duodenal atresia occurs between 1 in 1,000 and 1 in 5,000 live births. About 1/3 of infants born with duodenal atresia will also have Down Syndrome. Because of this association, newborns have often tested for other problems such as a heart defect.
What will happen during pregnancy?
 In general, duodenal atresia is difficult to diagnose during pregnancy. Prenatal diagnosis is usually based on non-specific signs on fetal ultrasound such as a dilated stomach. Because the amniotic fluid is normally swallowed and digested by the fetus, duodenal atresia can cause an increase in fluid in the amniotic sac, hydramnios. Although there are many other causes of hydramnios, this may be the first sign of a duodenal atresia.
In general, duodenal atresia is difficult to diagnose during pregnancy. Prenatal diagnosis is usually based on non-specific signs on fetal ultrasound such as a dilated stomach. Because the amniotic fluid is normally swallowed and digested by the fetus, duodenal atresia can cause an increase in fluid in the amniotic sac, hydramnios. Although there are many other causes of hydramnios, this may be the first sign of a duodenal atresia.
Duodenal atresia may be suspected by a routine prenatal ultrasound in the third trimester. Although the ultrasound may be suggestive that there is an abnormality, it cannot determine with 100% certainty that there is a bowel obstruction.
The obstetrician may order a special ultrasound that will examine the baby’s heart, also known as a fetal echocardiogram, and recommend an amniocentesis to look for chromosomal abnormalities. The mother’s amniotic fluid and the growth of the baby will be monitored closely with ultrasound by the obstetrician. Severe hydramnios may put the mother at risk for early delivery.
The fetal team will closely evaluate your fetus with duodenal atresia and help determine the best course of treatment. Your pregnancy will be closely monitored for complications. The Center coordinator will keep you in contact with the appropriate physicians and specialists as well as coordinating the care for you and your baby after delivery.
Will a fetal treatment be required?
 Although there are no prenatal treatment options for a baby with duodenal atresia, careful planning of delivery and care of the baby after birth can make a smooth transition for mother and child.
Although there are no prenatal treatment options for a baby with duodenal atresia, careful planning of delivery and care of the baby after birth can make a smooth transition for mother and child.
What special considerations should be made for delivery?
Type of delivery – Babies with duodenal atresia usually do not need a cesarian delivery. The delivery plan will be discussed with you and your obstetrician.
Place of delivery – As long as the baby does not demonstrate signs of distress, he or she can be cared for and delivered with usual obstetrical precautions. After birth, the baby can be safely transported to a treatment center with doctors and services such as a neonatal intensive care unit and the pediatric surgery. A child diagnosed with duodenal atresia will require an operation to address the problem and may stay in the hospital for several weeks.
Time of delivery -Intentional early delivery does not improve outcome. However, increasing amniotic fluid levels (hydramnios) does raise the chance for preterm delivery. If complications arise, the obstetrician may decide to induce delivery earlier than the expected due date.
http://childrens.memorialhermann.org/services/duodenal-atresia/
Duodenal atresia
Duodenal atresia occurs in the duodenum and causes a blockage. The duodenum is the bowel adjoining the stomach. Atresia means gap. Occasionally there may not be a complete atresia but a partial narrowing (stenosis) instead.
Duodenal atresia can be diagnosed on an ultrasound scan antenatally. This is a rare condition, the incidence is thought to be around 1 in 10,000 births. There is no known cause for this, but it is believed to have occurred sometime during the early weeks of pregnancy.
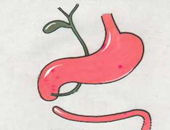
The diagnosis is made by seeing two fluid-filled areas in the baby’s abdomen which are the dilated stomach and duodenum. This is referred to as the ‘double bubble’ of duodenal atresia. This may have been detected on an antenatal scan or may not be detected until after the delivery when the baby starts to vomit.
Duodenal atresia can result in an increase in the amniotic fluid around the baby (known as polyhydramnios) which may lead to an early delivery.
Duodenal atresia can be associated with other abnormalities. Approximately one-third of babies with duodenal atresia have a chromosomal condition known as Down’s Syndrome and it is possible to test for this antenatally. This will be discussed with you by the team caring for you during your pregnancy. Other investigations may be necessary after the baby is born.
http://www.uhs.nhs.uk/OurServices/Childhealth/Neonatalsurgery/Conditionswetreat/DuodenalAtresia.aspx
Found a FaceBook Group dealing with this Support TEF/EA, Duodenal Atresia, CHD & g-tubes, Click to join.
Diagnosing Down Syndrome
Duodenal atresia is a birth defect of the digestive or gastrointestinal (GI) system that occurs more frequently in infants with Down Syndrome. Somewhere between 5 percent and 7 percent of infants with Down syndrome will be born with duodenal atresia, as compared to only 1 in 10,000 infants who do not have Down syndrome.
No one knows exactly why this happens, but it is known that it occurs early in the prenatal development of a fetus, prior to eleven weeks gestation. Rest assured that if your baby has duodenal atresia, there is nothing that you did to cause it or could have done to prevent it. Most infants with this problem do well after surgery.
https://www.verywell.com/duodenal-atresia-in-down-syndrome-1120476
Down Syndrome is typically diagnosed during pregnancy or shortly after birth. Learn about the screenings available and what they mean.
Step-by-Step Guide to Diagnosing Down Syndrome
Your pediatrician is often the first to suspect the diagnosis of Down syndrome in a newborn. The diagnosis is usually considered when a baby has certain physical findings, facial features, and possibly other birth defects. This step-by-step guide will explain what your pediatrician is looking for, and what tests are necessary to diagnose Down syndrome in a newborn baby. It is important to remember that while there are some similar findings that lead to a diagnosis of Down syndrome, no single baby with Down syndrome will have all of the features described here. Nor does the number of physical problems in a baby with Down syndrome correlate with their intellectual capacity. Each and every child with Down syndrome have their own unique personality and strengths.
https://www.verywell.com/making-the-diagnosis-of-down-syndrome-1120403
Are You at Risk of Having a Baby With Down Syndrome?
https://www.verywell.com/maternal-age-related-risks-for-down-syndrome-1120390
For the Full page on this click here
https://www.verywell.com/down-syndrome-diagnosis-4014232
For more info click here and lean, plus these are the support groups that can help you more.
Undescended Testicle Repair Surgery
What Is an Undescended Testicle Repair?
Undescended testicle repair surgery, also known as orchiopexy or orchidopexy, is a procedure that corrects cryptorchidism. This is a condition in which one or both testicles haven’t dropped into their proper position in the scrotum.
Orchiopexy is usually performed on infant males who are between 5 and 15 months old. The surgery reduces the risks of complications later in life.
Orchiopexy may also be performed on men whose undescended testicles weren’t corrected as children. However, the surgery isn’t recommended in all cases and should be discussed with a doctor.
The testicles begin developing in baby boys while they’re still inside their mother’s womb. Normally, the testicles drop down into the scrotum during the last few months before birth. In some cases, however, one or both testicles fail to descend correctly.
In half of these cases, a child’s testicles will drop down into their correct position within the scrotum within the first year of life without treatment. When the testicles don’t descend within the first year, the condition is known cryptorchidism. If your son has cryptorchidism, their doctor will likely recommend surgery to correct it. Undescended testicle repair surgery, also known as orchiopexy or orchidopexy, is an operation that’s commonly done to correct the placement of a testicle that hasn’t dropped into the scrotum. It’s usually performed on boys who are between 5 and 15 months old.
Why Is an Undescended Testicle Repair Performed?
Orchiopexy is performed to correct cryptorchidism, a condition in which one or both testicles haven’t descended into their proper position in the scrotum. If it’s left untreated, cryptorchidism can lead to infertility, increase the risk of testicular cancer, and cause hernias in the groin. It’s important to correct cryptorchidism in your child so that these risks are minimized.
Surgical options may differ for adult males whose undescended testicles weren’t corrected during childhood. Orchiopexy is usually the preferred choice for men who are age 32 and under. However, a doctor may suggest the complete removal of undescended testicles for younger men who are at a high risk of developing cancer. Orchiopexy usually isn’t performed on men over age 32, as there is an increased risk of adverse reactions to anesthesia. If you’re in this situation, consult with your doctor or a urologist to learn more about your options.
How Do We Prepare for an Undescended Testicle Repair?
Orchiopexy is done under general anesthesia, so certain rules for eating and drinking must be followed in the hours leading up to the procedure. The doctor will give your child specific instructions that they must follow.
While very young children may not realize that they’re going in for surgery, older children may get nervous before their procedure. They might feel especially nervous if you as a parent feel worried. Educate yourself about the procedure so that you feel comfortable and don’t unknowingly project your anxiety onto your son.
What Happens During an Undescended Testicle Repair?
Read more here
http://www.healthline.com/health/undescended-testicle-repair#Procedure4
http://www.surgeryencyclopedia.com/La-Pa/Orchiopexy.html
Cryptorchidism
Undescended Testicle
Cryptorchidism literally means hidden or obscure testis. It is synonymous with an incomplete testicular descent. The condition may be unilateral or bilateral. The term encompasses palpable, nonpalpable, and ectopic testicles The position of testis can be abdominal, inguinal, prescrotal, or gliding. Incidence is 3-5% in full-term boys, and 1.8% at one year of age.
The testicles descend to a scrotal position in human beings in order to optimise sperm production. The actual mechanisms of descent are unknown at present time. Certain important factors that cause proper descent include traction on testis by attachments in the scrotum, differential growth of the body wall, intra-abdominal pressure, maturation of the epididymis being responsible for migration of the testis. Multiple hormonal factors contribute also.
https://www.cornellurology.com/clinical-conditions/pediatric-urology/cryptorchidism/
And
http://www.beltina.org/health-dictionary/cryptorchidism-definition-treatment.html
What you read under here is to give you an idea about this rare condition, and a starting point from where you can start from.
Before you read what I have found and put together I would like to let you know of a Facebook Group I have stumbled across and in placing here above this post on my website the link is on the banner above. The group is called Skyler Journey with Feingold Syndrome.
What is Feingold syndrome?
Feingold syndrome is a disorder that affects many parts of the body. The signs and symptoms of this condition vary among affected individuals, even among members of the same family. Individuals with Feingold syndrome have characteristic abnormalities of their fingers and toes. Almost all people with this condition have a specific hand abnormality called brachymesophalangy, which refers to shortening of the second and fifth fingers. Other common abnormalities include fifth fingers that curve inward (clinodactyly), underdeveloped thumbs (thumb hypoplasia), and fusion (syndactyly) of the second and third toes or the fourth and fifth toes.
People with Feingold syndrome are frequently born with a blockage in part of their digestive system called gastrointestinal atresia. In most cases, the blockage occurs in the oesophagus (oesophagal atresia) or in part of the small intestine (duodenal atresia). Additional common features of Feingold syndrome include an unusually small head size (microcephaly), a small jaw (micrognathia), a narrow opening of the eyelids (short palpebral fissures), and mild to moderate learning disability. Less often, affected individuals have hearing loss, impaired growth, and kidney and heart abnormalities.
How common is Feingold syndrome?
Feingold syndrome appears to be a rare condition, although its exact prevalence is unknown.
What genes are related to Feingold syndrome?
Mutations in the MYCN gene cause Feingold syndrome. This gene provides instructions for making a protein that plays an important role in the formation of tissues and organs during embryonic development. Studies in animals suggest that this protein is necessary for normal development of the limbs, heart, kidneys, nervous system, digestive system, and lungs. The MYCN protein regulates the activity of other genes by attaching (binding) to specific regions of DNA. On the basis of this action, this protein is called a transcription factor.
Mutations in the MYCN gene that cause Feingold syndrome prevent one copy of the gene in each cell from producing any functional MYCN protein. As a result, only half the normal amount of this protein is available to control the activity of specific genes during embryonic development. It remains unclear how a reduced amount of the MYCN protein causes the specific features of Feingold syndrome.
What is the official name of the MYCN gene?
The official name of this gene is “v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog.” MYCN is the gene’s official symbol. The MYCN gene is also known by other names.
What is the normal function of the MYCN gene?
The MYCN gene provides instructions for making a protein that plays an important role in the formation of tissues and organs during embryonic development. Studies in animals suggest that this protein is necessary for normal development of the limbs, heart, kidneys, nervous system, digestive system, and lungs. The MYCN protein regulates the activity of other genes by attaching (binding) to specific regions of DNA. On the basis of this action, this protein is called a transcription factor.The MYCN gene belongs to a class of genes known as oncogenes. When mutated, oncogenes have the potential to cause normal cells to become cancerous. The MYCN gene is a member of the Myc family of oncogenes. These genes play important roles in regulating cell growth and division (proliferation) and the self-destruction of cells (apoptosis).
Does the MYCN gene share characteristics with other genes?
The MYCN gene belongs to a family of genes calledbHLH (basic helix-loop-helix). A gene family is a group of genes that share important characteristics. Classifying individual genes into families helps researchers describe how genes are related to each other.
Neuroblastoma – associated with the MYCN gene
Some gene mutations are acquired during a person’s lifetime and are present only in certain cells. These changes, which are not inherited, are called somatic mutations. Somatic mutations sometimes occur when DNA makes a copy of itself (replicates) in preparation for cell division. Errors in the replication process can result in one or more extra copies of a gene within a cell. The presence of extra copies of certain genes, known as gene amplification, can underlie the formation and growth of tumour cells. For example, amplification of the MYCN gene is found in about 25 percent of neuroblastomas. Neuroblastoma is a type of cancerous tumour that arises in developing nerve cells. The number of copies of the MYCN gene varies widely among these tumours but is typically between 50 and 100. Amplification of the MYCN gene is associated with a more severe form of neuroblastoma. It is unknown how amplification of this gene contributes to the aggressive nature of neuroblastoma.
How do people inherit Feingold syndrome?
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
Where can I find information about diagnosis or management of Feingold syndrome? JUST CLICK ON THESE LINKS
www.ncbi.nlm.nih.gov/books/NBK7050/
www.ncbi.nlm.nih.gov/gtr/conditions/C0796068/
www.ncbi.nlm.nih.gov/gtr/conditions/C3280489/
www.nlm.nih.gov/medlineplus/ency/article/003272.htm
Webbing of the fingers or toes
Other Name’s used Syndactyly; Polysyndactyly.
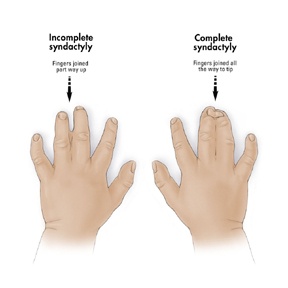
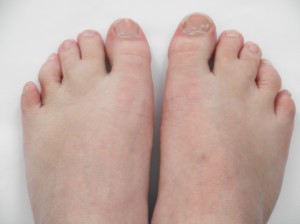
Feingold Syndrome.
Genetics Home Reference provides consumer-friendly information
https://ghr.nlm.nih.gov/condition/feingold-syndrome#
Feingold Syndrome 1
CLICK ON THE UNDERLINED RED WORDING TO TAKE YOU TO MORE INFO
Clinical characteristics.
Feingold syndrome 1 is characterized by digital anomalies (shortening of the 2nd and 5th middle phalanx of the hand, clinodactyly of the 5th finger, syndactyly of toes 2-3 and/or 4-5, thumb hypoplasia), microcephaly, facial dysmorphism (short palpebral fissures and micrognathia), gastrointestinal atresias (primarily esophageal and/or duodenal), and mild to moderate learning disability.
Diagnosis/testing.
Diagnosis is based on clinical findings. MYCN is the only gene in which mutation is known to cause Feingold syndrome 1.
Management.
Treatment of manifestations: Gastrointestinal atresia is treated surgically. Hearing loss, renal anomalies, and cardiac anomalies are treated in the usual manner.
Genetic counseling.
Feingold syndrome 1 is inherited in an autosomal dominant manner. Approximately 60% of individuals with Feingold syndrome 1 have an affected parent; the proportion of cases caused by de novo mutations is unknown. Each child of an individual with Feingold syndrome 1 has a 50% chance of inheriting the mutation. Prenatal diagnosis for pregnancies at increased risk is possible if the disease-causing mutation in the family has been identified.
For more info click on this link below, and work your way through the mass of links.
jackhammer Esophagus
New minimally-invasive procedure to treat jackhammer oesophagus
Six weeks ago 67-year old John Galbreath welcomed death, rather than live one more day. Intense pain in his chest had left him nearly bedridden for four months. Galbreath had a condition called jackhammer oesophagus, which made him feel like the name suggests—like a jackhammer was going off in his chest.
“This is a condition where the oesophagus spasms out of control and causes the patient to feel like they are having a heart attack 24/7,” said Dr Brian Dunkin, a minimally invasive surgeon at Houston Methodist Hospital. “With this condition, the oesophagus does not contract properly causing patients to either have terrible chest pain or not be able to swallow food or fluid without it backing up into the oesophagus. Now there’s an easier way to fix the problem, and we are the only ones doing it in Texas.”
The standard treatment for jackhammer oesophagus is to perform surgery through the chest and can include up to five incisions. Sometimes a tube is placed in the chest for two to three days and there is a minimum of two weeks off work.Now there’s a new minimally-invasive procedure to treat jackhammer oesophagus and another disorder called achalasia, which is when the opening between the oesophagus and stomach is blocked.
The procedure, Per Oral Endoscopic Myotomy or POEM, involves fixing the problems through the mouth instead of the chest. Like they did with Galbreath, surgeons operate through the mouth using a gastroscope, a thin flexible camera made to look into the oesophagus and stomach. They drive the gastroscope down under the mucosa, the inner lining of the oesophagus, all the way to the stomach wall and then cut the muscle of the lower oesophagus and upper stomach to relieve the spasm and blockage. The POEM procedure leaves no surgical scars and is essentially pain-free and patients can return to work in less than a week.
“Most of these patients don’t realise how bad their situation has gotten until they can swallow again,” Dunkin said. “The pain and heartburn from spasm or food and fluid backing up are virtually gone and they can get on with a normal life.”
Galbreath says he felt like a new man as soon as he woke up from surgery.
“On Super Bowl Sunday, I was able to eat shrimp, chips and a hot dog with no pain or discomfort,” Galbreath said. “After years of being misdiagnosed, I cannot tell you how happy I am that I was properly diagnosed and able to undergo this procedure.”
Source: Houston Methodist Hospital
https://www.verywell.com/what-is-jackhammer-esophagus-1191896
.
Hypertensive peristalsis
Hypertensive peristalsis, also known as nutcracker oesophagus, and or Esophageal Spasm is diagnosed when contractions proceed in a coordinated manner but the amplitude is excessive. Hypercontractile oesophagus, also known as jackhammer oesophagus, is an extreme phenotype of hypertensive contractions in which contractions are of very high amplitude, involving a majority of the oesophagus, and whose duration occurs for a prolonged period with a jackhammer-type appearance on high-resolution manometry.
Symptoms can include dysphagia, regurgitation, and noncardiac chest pain. Because of the vague symptoms and difficulty in diagnosis, the oesophagal spasm is often underdiagnosed and therefore not adequately treated. In many patients, manometric and radiologic abnormalities may not correlate with symptom presentation.
Currently, high-resolution manometry is the best diagnostic modality. Treatment includes calcium channel blockers, botulinum toxin, nitrates, tricyclic antidepressants, sildenafil, dilatation, myotomy, and esophagectomy. Research is ongoing to determine the underlying causes to improve diagnostic capabilities and therapeutic regimens in the future.
http://emedicine.medscape.com/article/174975-overview
What is an esophageal manometry test?
An esophageal manometry test is a test used to evaluate the function of the esophagus and the lower esophageal sphincter (LES). It must be well understood that this is not a specific test for GERD. Rather, as the function is assessed, your physician may see changes that are consistent with GERD or may also discover specific esophageal contraction disorders that may mimic GERD. Approximately 5% of people thought to have GERD actually have one of the several esophageal disorders uncovered with an esophageal manometry test. Also, many proven to have GERD by previous pH testing have normal or close to normal manometry studies.
To understand what happens during the manometry test, you must understand what happens when you swallow. In order to pass food, the esophagus must contract in a sequential manner after swallowing. The top end must contract first, then the middle, and then the bottom. This propels the food downward toward the stomach. At the same time, the LES, located at the end of the esophagus and just above the stomach, must open or relax to allow the food to pass into the stomach. All of these events are measured during manometry test in terms of pressure. As part of the esophagus squeezes, it generates pressure. As the LES opens, its pressure lowers from its resting state. The pressure measurements also measure the length of the LES and can detect a hiatus hernia, if present.
How is the test conducted?
The study, which takes about 20 minutes to complete, is conducted by passing a thin flexible tube containing 36 separate pressure sensors gently through the nose into the stomach. During this test, you will be awake, but the nose is numbed with topical medicine. Once the tube is in place, you will be given a small amount of water to swallow. As you swallow, the pressure measurements are processed by a computer and displayed on a screen. After 10 swallows, the test is concluded and the tube is removed. You will immediately be able to return to normal diet and activities.
As noted above, this is not a test for GERD. A pH study usually is required for that. However, in many patients with GERD, a manometry test can reveal important information about your condition. A manometry test can also diagnose other conditions of the esophagus. Two examples include:
https://www.verywellhealth.com/esophageal-manometry-test-4157830
Click the highlighted Words to be taken to pages to explain.
Achalasia: With this condition, the LES fails to relax. As a result, food cannot pass into the stomach and the patient will experience difficulty swallowing.
Nutcracker esophagus: ( also known as Jackhammer Esophagus.) This occurs when the esophagus squeezes in sequence, but too vigorously. This can cause chest pain, as well as swallowing difficulty.
Iron-Deficiency Anemia
Anemia is a condition in which the body does not have enough healthy red blood cells. Iron helps make red blood cells. When your body does not have enough iron, it will make fewer red blood cells or red blood cells that are too small. This is called iron deficiency anemia.
causes
Iron deficiency anemia is the most common form. Red blood cells bring oxygen to the body’s tissues. Healthy red blood cells are made in your bone marrow. Red blood cells circulate through your body for 3 to 4 months. Parts of your body like your spleen remove old blood cells. Iron is a key part of red blood cells. Without iron, the blood cannot carry oxygen effectively. Your body normally gets iron through your diet. It also reuses iron from old red blood cells.
You get iron deficiency anemia when your body’s iron stores run low.
This can occur because:
http://www.nytimes.com/health/guides/disease/anemia/overview.html
Click the underlined Blue Writing
Iron-deficiency anemia is a common, easily treated condition that occurs if you don’t have enough iron in your body. Low iron levels usually are due to blood loss, poor diet, or an inability to absorb enough iron from food.
Routine Iron Supplementation and Screening for Iron Deficiency Anemia in Children Ages 6 to 24 Months
This holds a lot of info for Children.
http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0073392/


